
Unten eingestellt ist eine HSP-Forschungsarbeit, die sich mit dem Protein REEP2 befasst. Zunächst sei darauf hingewiesen, dass dieses Protein aus einem Gen entwickelt wird, das die Nummer 72 in der Liste der HSP-Gene führt. Beim Gen handelt es sich also um das SPG72. Das REEP2 gehört zu einer Gruppe von Proteinen, die REEP1 bis REEP6 genannt werden. Bisher kannten wir nur das REEP1 als ein Protein, das mit der HSP in Zusammenhang steht. Das REEP1 wird bekanntlich durch das Gen SPG31 gebildet. Die Familie der REEP-Proteine ist für die korrekte Steuerung anderer Proteine, der so genannten G-Proteine verantwortlich sind. REEP steht als Abkürzung für „receptor expressing enhancing proteins“.
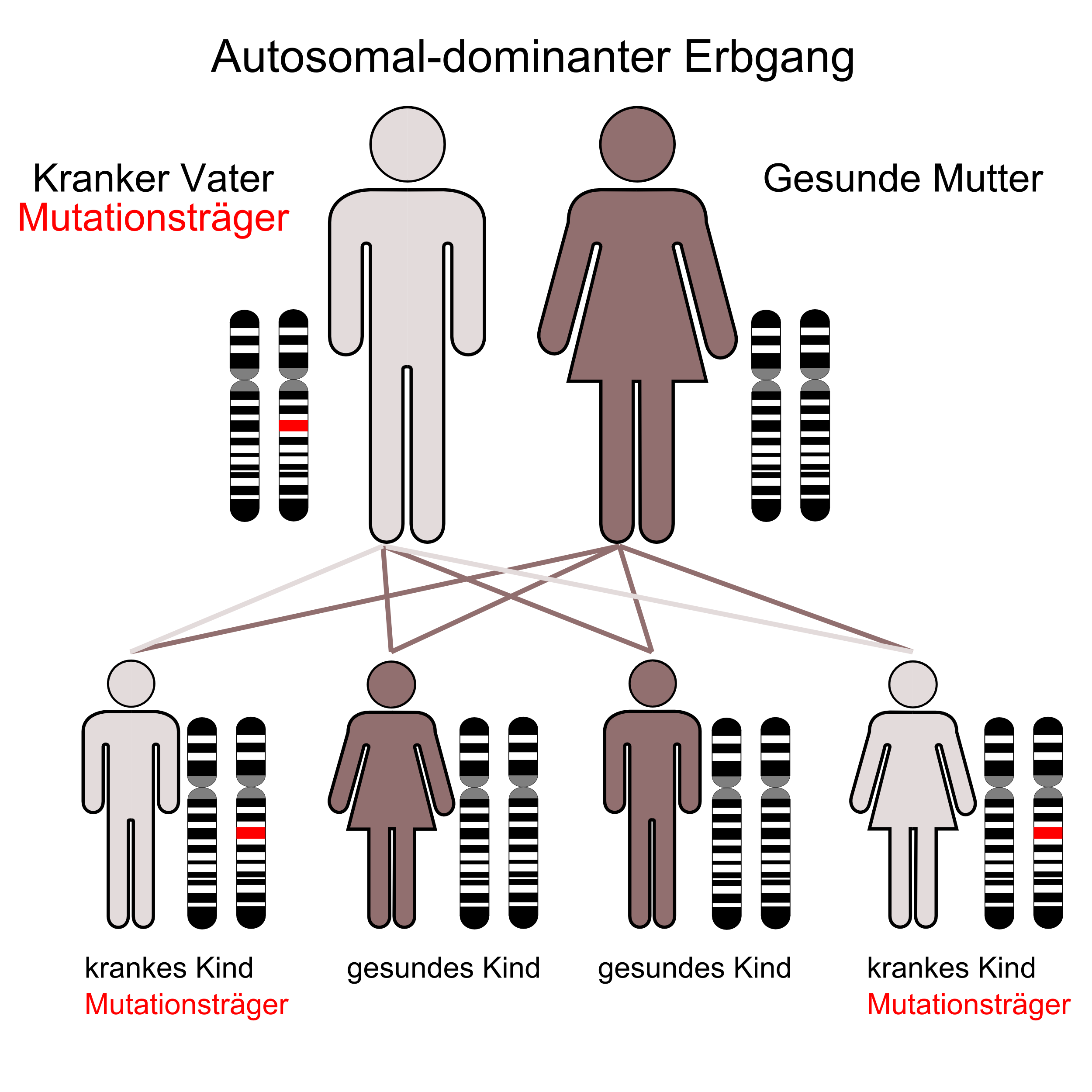
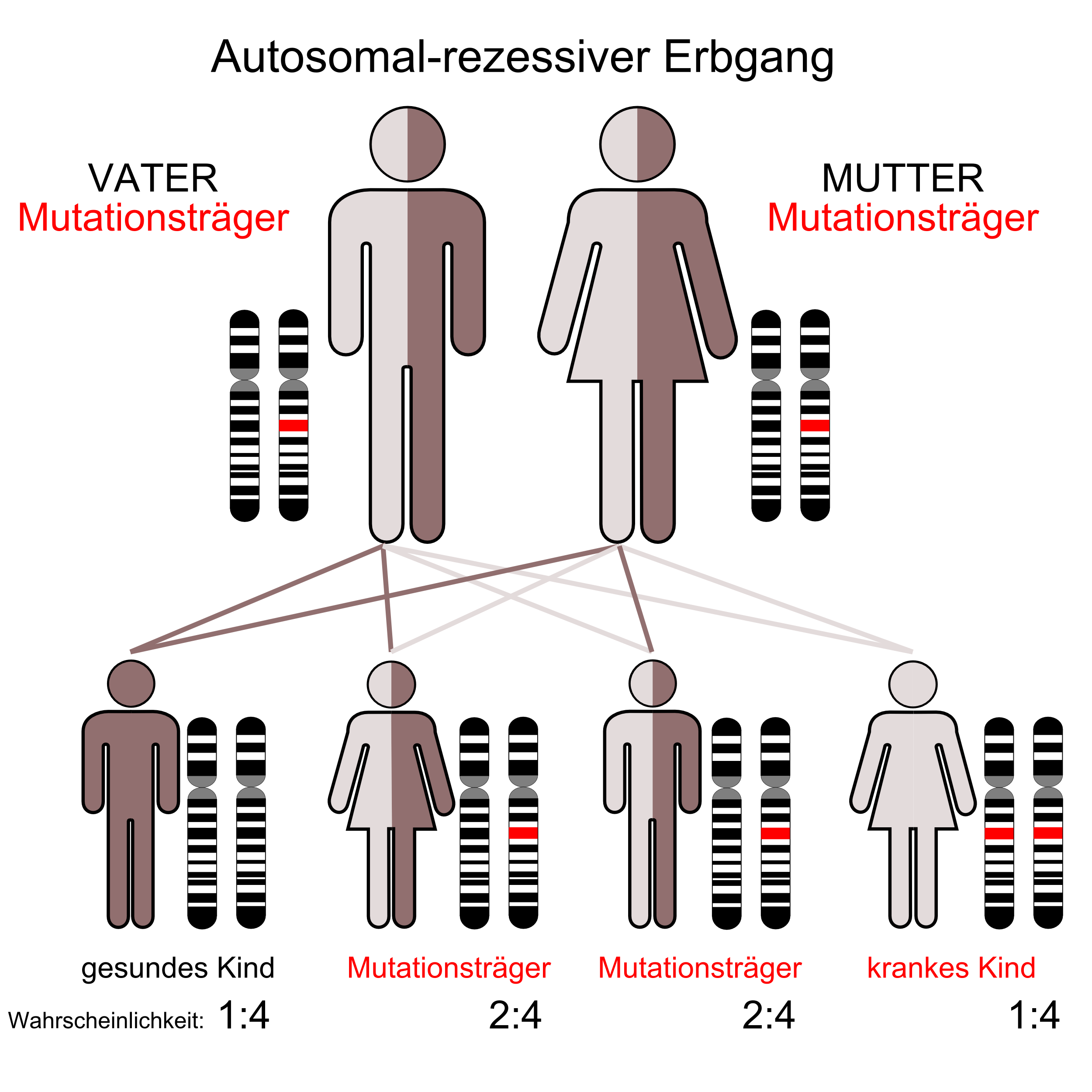
Das Besondere beim REEP2 ist es, dass das zugehörige Gen SPG72 sowohl autosomal dominant, wie auch autosomal rezessiv aktiv sein kann. Das klingt im ersten Moment unlogisch, weil wir bisher der Meinung waren, dass ein Gen entweder dominant oder rezessiv ist. Genauere Infos zum Gen sind hier zu finden. Anfang diesen Jahres gab es bereits eine andere Publikation, in der das gleiche Ergebnis beim Gen SPG3A (Protein Atlastin1) beschrieben wurde. Die entsprechende Veröffentlichung ist ebenfalls unten eingestellt.
{kind=link}
{kind=link}
Beide Arbeiten zeigen, dass die bisher so klare Zuordnung zu „autosomal dominant“ und „autosomal rezessiv“ nicht mehr in jedem Fall gültig zu sein scheint. Deshalb hat diese Erkenntnis große Auswirkungen sowohl auf die medizinische Gendiagnostik wie auch auf die private Familienplanung. Es ist erkennbar, dass durch solche Ergebnisse aus der Grundlagenforschung, das Bild zur HSP immer komplexer und umfangreicher wird. Dabei wird es leider nicht einfacher. Es wird sich zeigen, ob es weitere HSP-Gene gibt, die sowohl dominant wie auch rezessiv aktiv werden können. Sollte es weitere Nachrichten in diesem Zusammenhang geben, dann werden die natürlich auch hier einstellen, weil sie für jeden HSP-Erkrankten persönlich wichtig sein können.
——————————————–
6. Feb 2014
Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia.
Esteves T, Durr A, Mundwiller E, Loureiro JL, Boutry M, Gonzalez MA, Gauthier J, El-Hachimi KH, Depienne C, Muriel MP, Acosta Lebrigio RF, Gaussen M, Noreau A, Speziani F, Dionne-Laporte A, Deleuze JF, Dion P, Coutinho P, Rouleau GA, Zuchner S, Brice A, Stevanin G, Darios F.
Hereditary spastic paraplegias (HSPs) are clinically and genetically heterogeneous neurological conditions. Their main pathogenic mechanisms are thought to involve alterations in endomembrane trafficking, mitochondrial function, and lipid metabolism. With a combination of whole-genome mapping and exome sequencing, we identified three mutations in REEP2 in two families with HSP: a missense variant (c.107T>A [p.Val36Glu]) that segregated in the heterozygous state in a family with autosomal-dominant inheritance and a missense change (c.215T>A [p.Phe72Tyr]) that segregated in trans with a splice site mutation (c.105+3G>T) in a family with autosomal-recessive transmission. REEP2 belongs to a family of proteins that shape the endoplasmic reticulum, an organelle that was altered in fibroblasts from an affected subject. In vitro, the p.Val36Glu variant in the autosomal-dominant family had a dominant-negative effect; it inhibited the normal binding of wild-type REEP2 to membranes. The missense substitution p.Phe72Tyr, in the recessive family, decreased the affinity of the mutant protein for membranes that, together with the splice site mutation, is expected to cause complete loss of REEP2 function. Our findings illustrate how dominant and recessive inheritance can be explained by the effects and nature of mutations in the same gene. They have also important implications for genetic diagnosis and counseling in clinical practice because of the association of various modes of inheritance to this new clinico-genetic entity.
Quelle: http://www.ncbi.nlm.nih.gov/pubmed/24388663
———————————————————————————————-
29. Jan 2014
Evidence for autosomal recessive inheritance in SPG3A
caused by homozygosity for a novel ATL1 missense mutation.
Khan TN, Klar J, Tariq M, Anjum Baig S, Malik NA, Yousaf R, Baig SM, Dahl N.
Hereditary spastic paraplegias (HSPs) comprise a heterogeneous group of disorders characterized by progressive spasticity and weakness of the lower limbs. Autosomal dominant and ‚pure‘ forms of HSP account for ∼80% of cases in Western societies of whom 10% carry atlastin-1 (ATL1) gene mutations. We report on a large consanguineous family segregating six members with early onset HSP. The pedigree was compatible with both autosomal dominant and autosomal recessive inheritance. Whole-exome sequencing and segregation analysis revealed a homozygous novel missense variant c.353G>A, p.(Arg118Gln) in ATL1 in all six affected family members. Seven heterozygous carriers, five females and two males, showed no clinical signs of HSP with the exception of sub-clinically reduced vibration sensation in one adult female. Our combined findings show that homozygosity for the ATL1 missense variant remains the only plausible cause of HSP, whereas heterozygous carriers are asymptomatic. This apparent autosomal recessive inheritance adds to the clinical complexity of spastic paraplegia 3A and calls for caution using directed genetic screening in HSP.